有机光电材料与器件的迅速发展得益于过渡金属催化的C−X/C−M传统偶联反应的发展。近年来,C−H键活化被誉为有机合成化学的“圣杯”,备受关注,但利用C−H键活化构筑有机功能分子的工作却刚刚起步,其研究还远未受到重视。化学家越来越意识到利用C−H键活化构筑有机功能材料的重要性。
含(苯并)噻吩的联杂芳烃骨架是有机光电材料中广泛存在的结构片段。过渡金属催化的杂芳烃之间的氧化C−H/C−H偶联反应无疑是构筑该类骨架最为简洁高效的策略之一。然而,由于(苯并)噻吩自身C2和C3位的电性差异,无论经历芳基亲电取代(SEAr)或协同脱氢金属化(CMD)的机制,(苯并)噻吩的C−H/C−H型芳基化反应均发生在C2位,因此实现(苯并)噻吩选择性的C3位C−H/C−H芳基化反应面临巨大挑战。

图1.(苯并)噻吩与(杂)芳烃氧化C−H/C−H交叉偶联反应发展历程
针对这一问题,游劲松课题组通过改变催化中心金属亲电性来扭转苯并噻吩在C−H/C−H氧化偶联反应中的区域选择性,实现了首例苯并噻吩C3位区域选择性C−H/C−H芳基化反应,并成功用于开发非传统结构的OLED新材料。机理研究表明,三氟甲磺酸根对实现C3位选择性活化具有重要作用,此外作者还成功分离得到苯并噻吩加成的七元环铑去芳构化中间体,晶体结构证明该中间体需经历syn β-H消除才能得到联杂芳烃产物。底物拓展方面,该反应展现出了良好的官能团兼容性,含各类供吸电子基团(烷基、甲氧基、卤素、三氟甲基、酰基、酯基、硝基等)和更大p共轭度的底物均能在该催化体系下顺利转化为目标产物。偶联产物经导向基脱除、分子内环化等衍生还能够顺利得到[4]螺烯构型的苯并噻吩并异喹啉酮骨架以及苯并噻吩并异喹啉。
通过该合成方法,作者高效构筑了一类具有非传统结构的新型受体(2,3-c-BTIQO),相比于C2位连接的线性平面受体结构,其独特的螺烯构型赋予发光分子热活化延迟荧光(TADF)性质,并成功用于制备高效率蓝光OLED器件,器件发光波长为472 nm,最大外量子效率为25.4%。
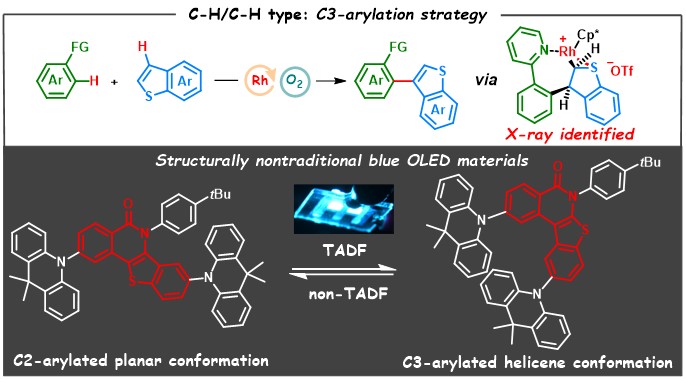
图2. 苯并噻吩C3位选择性芳基化反应及OLED材料设计思路
该研究以“Insight into Regioselective Control in Aerobic Oxidative C–H/C–H Coupling for C3-Arylation of Benzothiophenes: Toward Structurally Nontraditional OLED Materials”为题目发表在Journal of the American Chemical Society上,四川大学为第一单位,bwin必赢游劲松教授和宾正杨副研究员为该论文通讯作者,师洋博士为论文的第一作者。特别感谢国家自然科学基金委、四川省科技厅、四川大学的经费支持。
文章链接: https://pubs.acs.org/doi/pdf/10.1021/jacs.1c11277
